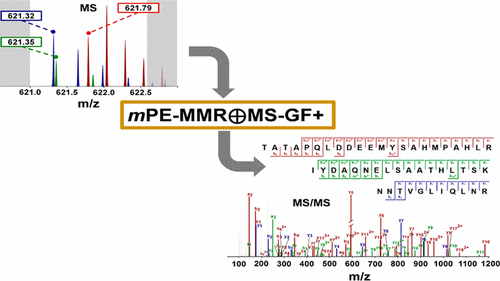
Mass spectrometry (MS)-based proteomics, which uses high-resolution hybrid mass spectrometers such as the quadrupole-orbitrap mass spectrometer, can yield tens of thousands of tandem mass (MS/MS) spectra of high resolution during a routine bottom-up experiment. Despite being a fundamental and key step in MS-based proteomics, the accurate determination and assignment of precursor monoisotopic masses to the MS/MS spectra remains difficult. The difficulties stem from imperfect isotopic envelopes of precursor ions, inaccurate charge states for precursor ions, and cofragmentation. We describe a composite method of utilizing MS data to assign accurate monoisotopic masses to MS/MS spectra, including those subject to cofragmentation. The method, “multiplexed post-experiment monoisotopic mass refinement” (mPE-MMR), consists of the following: multiplexing of precursor masses to assign multiple monoisotopic masses of cofragmented peptides to the corresponding multiplexed MS/MS spectra, multiplexing of charge states to assign correct charges to the precursor ions of MS/MS spectra with no charge information, and mass correction for inaccurate monoisotopic peak picking. When combined with MS-GF+, a database search algorithm based on fragment mass difference, mPE-MMR effectively increases both sensitivity and accuracy in peptide identification from complex high-throughput proteomics data compared to conventional methods.
http://pubs.acs.org/doi/abs/10.1021/acs.analchem.6b03874