The vibrational frequency of a chosen normal mode is one of the most accurately measurable spectroscopic properties of molecules in condensed phases. Accordingly, infrared absorption and Raman scattering spectroscopy have provided valuable information on both distributions and ensemble-average values of molecular vibrational frequencies, and these frequencies are now routinely used to investigate structure, conformation, and even absolute configuration of chemical and biological molecules of interest. Recent advancements in coherent time-domain nonlinear vibrational spectroscopy have allowed the study of heterogeneous distributions of local structures and thermally driven ultrafast fluctuations of vibrational frequencies. To fully utilize IR probe functional groups for quantitative bioassays, a variety of biological and chemical techniques have been developed to site-specifically introduce vibrational probe groups into proteins and nucleic acids. These IR-probe-labeled biomolecules and chemically reactive systems are subject to linear and nonlinear vibrational spectroscopic investigations and provide information on the local electric field, conformational changes, site–site protein contacts, and/or function-defining features of biomolecules.
A rapidly expanding library of data from such experiments requires an interpretive method with atom-level chemical accuracy. However, despite prolonged efforts to develop an all-encompassing theory for describing vibrational solvatochromism and electrochromism as well as dynamic fluctuations of instantaneous vibrational frequencies, purely empirical and highly approximate theoretical models have often been used to interpret experimental results. They are, in many cases, based on the simple assumption that the vibrational frequency of an IR reporter is solely dictated by electric potential or field distribution around the vibrational chromophore. Such simplified description of vibrational solvatochromism generally referred to as vibrational Stark effect theory has been considered to be quite appealing and, even in some cases, e.g., carbonyl stretch modes in amide, ester, ketone, and carbonate compounds or proteins, it works quantitatively well, which makes it highly useful in determining the strength of local electric field around the IR chromophore. However, noting that the vibrational frequency shift results from changes of solute–solvent intermolecular interaction potential along its normal coordinate, Pauli exclusion repulsion, polarization, charge transfer, and dispersion interactions, in addition to the electrostatic interaction between distributed charges of both vibrational chromophore and solvent molecules, are to be properly included in the theoretical description of vibrational solvatochromism. Since the electrostatic and nonelectrostatic intermolecular interaction components have distinctively different distance and orientation dependences, they affect the solvatochromic vibrational properties in a completely different manner.
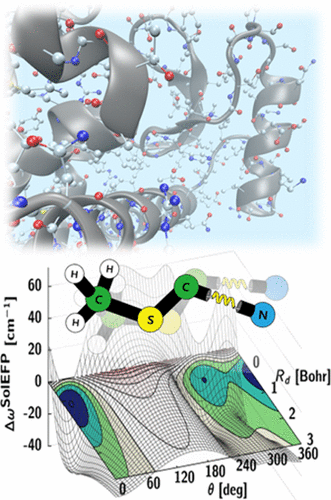
Over the past few years, we have developed a systematic approach to simulating vibrational solvatochromic data based on the effective fragment potential approach, one of the most accurate and rigorous theories on intermolecular interactions. We have further elucidated the interplay of local electric field with the general vibrational solvatochromism of small IR probes in either solvents or complicated biological systems, with emphasis on contributions from non-Coulombic intermolecular interactions to vibrational frequency shifts and fluctuations. With its rigorous foundation and close relation to quantitative interpretation of experimental data, this and related theoretical approaches and experiments will be of use in studying and quantifying the structure and dynamics of biomolecules with unprecedented time and spatial resolution when combined with time-resolved vibrational spectroscopy and chemically sensitive vibrational imaging techniques.